Developing a new drug from original idea to the launch of a finished product is a complex process which can take 12–15 years and cost in excess of $1 billion. A drug discovery programme initiates because there is a disease or clinical condition without suitable medical products available and it is this unmet clinical need which is the underlying driving motivation for the project.
During lead discovery, an intensive search ensues to find a drug-like small molecule or biological therapeutic, typically termed a development candidate, that will progress into preclinical, and if successful, into clinical development and ultimately be a marketed medicine. There are five critical steps in the U.S. FDA drug development process, including many phases and stages within each of them.
Step 1: Discovery and Development
Step 2: Preclinical Research
Step 3: Clinical Development
Step 4: FDA Review
Step 5: FDA Post-Market Safety Monitoring (Pharmacovigilance)
Pharmacovigilance certification and drug safety training learn from Company Connect Consultancy

Discovery of New Drug:
Discovery of new drug takes place via,
1. New insights into a disease process that allow researchers to design a product to stop or reverse the effects of the disease.
2. Many tests of molecular compounds to find possible beneficial effects against any of a large number of diseases.
3. Existing treatments that have unanticipated effects.
4. New technologies, such as those that provide new ways to target medical products to specific sites within the body or to manipulate genetic material.
At this stage, thousands of compounds may be potential candidates for development as a medical treatment. After early testing, however, only a small number of compounds look promising and call for further study.
Development of New Drug:
Once a perfect compound is identified, researchers conduct experiments to gather further information’s on:
1. How the compound is absorbed, distributed, metabolized, and excreted.
2. Its potential benefits and mechanisms of action.
3. The best dosage.
4. The best way to give the drug (such as by mouth or injection).
5. Side effects or adverse events that can often be referred to as toxicity.
6. How it affects different groups of people (such as by gender, race, or ethnicity) differently.
7. How it interacts with other drugs and treatments.
Its effectiveness as compared with similar drugs.

Target Identification and Validation:
Target identification finds a gene or protein (therapeutic agent) that plays a significant role in disease. Afterward, scientists and researchers record the target’s therapeutic characteristics. Drug targets must be efficacious, safe, usable, and capable of meeting clinical and commercial requirements. To validate targets, researchers use modern tools and techniques such as disease association, bioactive molecules, cell-based models, protein interactions, signalling pathways analysis, functional analysis of genes, in vitro genetic manipulation, antibodies, and chemical genomics. For example, the Sanger Whole Genome CRISPER library and Duolink PLA are excellent sources for drug discovery targets.
Hit Discovery Process:
Following target validation, compound screening assays are developed.
Assay Development and Screening:
Assay development in drug discovery is a crucial component of drug discovery workflow. Assays are test systems that evaluate the effects of the new drug candidate at the cellular, molecular, and biochemical levels.
High Throughput Screening:
High Throughput Screening (HTS) uses robotics, data processing/control software, liquid handling devices, and sensitive detectors to rapidly conduct millions of pharmacological, chemical, and genetic tests, eliminating hours of painstaking testing by scientists. HTS identifies active compounds, genes, or antibodies that affect human molecules.
Hit To Lead:
In the Hit to Lead (H2L) process, small molecule hits from an HTS are evaluated and optimized in a limited way into lead compounds. These compounds then move on to the lead optimization process.
Lead Optimization:
In the lead optimization (LO) process, the lead compounds discovered in the H2L process are synthesized and modified to improve potency and reduce side effects. Lead optimization conducts experimental testing using animal efficacy models and ADMET tools, designing the drug candidate.
Active Pharmaceutical Ingredients:
Active pharmaceutical ingredients (APIs) are biologically active ingredients in a drug candidate that produce desired effects. All drugs are made up of the API or APIs and excipients. Excipients are inactive substances that deliver the drug into the human system. High Potency Active Pharmaceutical Ingredients (HP APIs) are molecules that are effective at much smaller dosage levels than standard APIs. They are classified based on toxicity, pharmacological potency, and occupational exposure limits (OELs), and used in complex drug development involving more than ten steps.
Preclinical Research:
Before testing a drug in people, researchers must find out whether it has the potential to cause serious harm, also called toxicity. The two types of preclinical research are:
1. In-Vivo
2. In-Vitro
FDA requires researchers to use good laboratory practices (GLP), defined in medical product development regulations, for preclinical laboratory studies. The GLP regulations are found in 21 CFR Part 58.1: Good Laboratory Practice for Nonclinical Laboratory Studies. These regulations set the minimum basic requirements for:
1. Study conduct
2. personnel
3. facilities
4. equipment
5. written protocols
6. operating procedures
7. study reports
8. and a system of quality assurance oversight for each study to help assure the safety of FDA-regulated product.
Usually, preclinical studies are not very large. However, these studies must provide detailed information on dosing and toxicity levels. After preclinical testing, researchers review their findings and decide whether the drug should be tested in people.
Preclinical Trials test the new drug on non-human subjects for efficacy, toxicity, and pharmacokinetic (PK) information. The following figure depicts the preclinical trails study:

Clinical Drug Development process and it’s Online Training Clinical Research:
Once preclinical research is complete, researchers move on to clinical drug development, including clinical trials and volunteer studies to finetune the drug for human use. While preclinical research answers basic questions about a drug’s safety, it is not a substitute for studies of ways the drug will interact with the human body. “Clinical research” refers to studies, or trials, that are done in people. As the developers design the clinical study, they will consider what they want to accomplish for each of the different Clinical Research Phases and begin the Investigational New Drug Process (IND). A process they must go through before clinical research begins:
1. Complexity Of Study Design, Associated Cost & Implementation Issues.
2. Clinical Trials– Dose Escalation, Single Ascending & Multiple Dose Studies.
Best Institute for Pharmacovigilance and Clinical Research training Course from Company Connect Consultancy
Process of Clinical research can be better understood by the following picture:

Designing Clinical Trials:
Researchers design clinical trials to answer specific research questions related to a medical product. These trials follow a specific study plan, called a protocol, that is developed by the researcher or manufacturer. Before a clinical trial begins, researchers review prior information about the drug to develop research questions and objectives. Then, they decide:
1. Who qualifies to participate (selection criteria)
2. How many people will be part of the study?
3. How long the study will last
4. Whether there will be a control group and other ways to limit research bias
5. How the drug will be given to patients and at what dosage
6. What assessments will be conducted, when, and what data will be collected
7. How the data will be reviewed and analyzed
Clinical trials follow a typical series from early, small-scale, Phase 1 studies to late-stage, large scale, Phase 3 studies.
Investigational New Drug Process:
Drug developers, or sponsors, must submit an Investigational New Drug (IND) application to FDA before beginning clinical research.
In the IND application, developers must include:
1. Animal study data and toxicity (side effects that cause great harm) data
2. Manufacturing information
3. Clinical protocols (study plans) for studies to be conducted
4. Data from any prior human research
5. Information about the investigator.
Clinical Research Phase Studies:
Clinical research takes place in following four phases.

Asking for FDA Assistance:
Drug developers are free to ask for help from FDA at any point in the drug development process, including:
1. Pre-IND application, to review FDA guidance documents and get answers to questions that may help enhance their research
2. After Phase 2, to obtain guidance on the design of large Phase 3 studies
3. Any time during the process, to obtain an assessment of the IND application
As long as clinical trials are thoughtfully designed, reflect what developers know about a product, safeguard participants, and otherwise meet Federal standards, FDA allows wide latitude in clinical trial design.
FDA IND Review:
The review team consists of a group of specialists in different scientific fields. Each member has different responsibilities. Once the new drug has been formulated for its best efficacy and safety, and the results from clinical trials are available, it is forwarded for holistic FDA review. At this time, the FDA reviews and approves, or does not approve, the drug application submitted by the drug development company.
Approval:
The FDA review team has 30 days to review the original IND submission. The process protects volunteers who participate in clinical trials from unreasonable and significant risk in clinical trials. FDA responds to IND applications in one of two ways:
Approval to begin clinical trials.
Clinical hold to delay or stop the investigation. FDA can place a clinical hold for specific reasons, including:
Participants are exposed to unreasonable or significant risk.
Investigators are not qualified.
Materials for the volunteer participants are misleading.
The IND application does not include enough information about the trial’s risks.
A clinical hold is rare; instead, FDA often provides comments intended to improve the quality of a clinical trial. In most cases, if FDA is satisfied that the trial meets Federal standards, the applicant is allowed to proceed with the proposed study.
The developer is responsible for informing the review team about new protocols, as well as serious side effects seen during the trial. This information ensures that the team can monitor the trials carefully for signs of any problems. After the trial ends, researchers must submit study reports.
This process continues until the developer decides to end clinical trials or files a marketing application. Before filing a marketing application, a developer must have adequate data from two large, controlled clinical trials.
FDA Review (Learn Regulatory affairs course online)
If a drug developer has evidence from its early tests and preclinical and clinical research that a drug is safe and effective for its intended use, the company can file an application to market the drug. The FDA review team thoroughly examines all submitted data on the drug and makes a decision to approve or not to approve it.
New Drug Application:
A New Drug Application (NDA) tells the full story of a drug. Its purpose is to demonstrate that a drug is safe and effective for its intended use in the population studied.
A drug developer must include everything about a drug—from preclinical data to Phase 3 trial data—in an NDA. Developers must include reports on all studies, data, and analyses. Along with clinical results, developers must include:
1. Proposed labelling
2. Safety updates
3. Drug abuse information
4. Patent information
5. Any data from studies that may have been conducted outside the United States
6. Institutional review board compliance information
7. Directions for use
FDA Review:
Once FDA receives an NDA, the review team decides if it is complete. If it is not complete, the review team can refuse to file the NDA. If it is complete, the review team has 6 to 10 months to make a decision on whether to approve the drug.
FDA Approval:
In cases where FDA determines that a drug has been shown to be safe and effective for its intended use, it is then necessary to work with the applicant to develop and refine prescribing information. This is referred to as “labelling.” Labelling accurately and objectively describes the basis for approval and how best to use the drug.
Often, though, remaining issues need to be resolved before the drug can be approved for marketing. Sometimes FDA requires the developer to address questions based on existing data. In other cases, FDA requires additional studies. At this point, the developer can decide whether or not to continue further development. If a developer disagrees with an FDA decision, there are mechanisms for formal appeal.
FDA Advisory Committees:
Often, the NDA contains sufficient data for FDA to determine the safety and effectiveness of a drug. Sometimes, though, questions arise that require additional consideration. In these cases, FDA may organize a meeting of one of its Advisory Committees to get independent, expert advice and to permit the public to make comments. These Advisory Committees include a Patient Representative that provides input from the patient perspective. Learn more about FDA Advisory Committees.
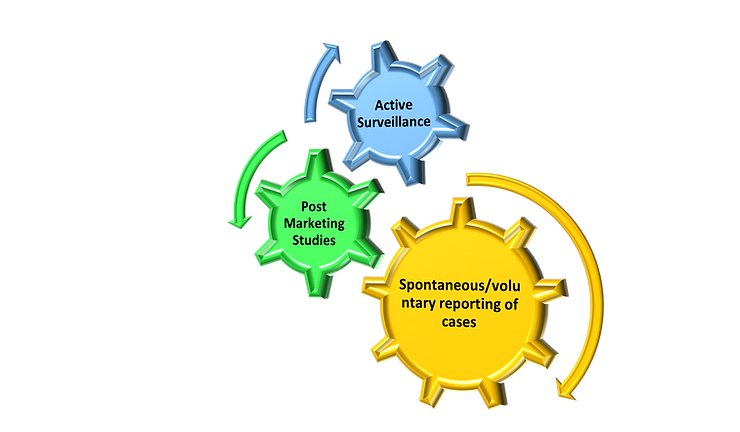
FDA Post-Market Drug Safety Monitoring: Pharmacovigilance training at Hyderabad and Bangalore
Following drug approval and manufacturing, the FDA requires drug companies to monitor the safety of its drug using the FDA Adverse Event Reporting System (FAERS) database. FAERS helps FDA implement its post-marketing safety surveillance program. Through this program, manufacturers, health professionals, and consumers report problems with approved drugs.
The FDA requires drug monitoring since clinical trials provide important information on a drug’s efficacy and safety, it is impossible to have complete information about the safety of a drug at the time of approval.